 SBARS.IMPB.RU
en |
SBARS.IMPB.RU
en | LSCGAT: Long Sequences Customizable Global Alignment Tool.
Guide
The capabilities of the application can be demonstrated in the following example.
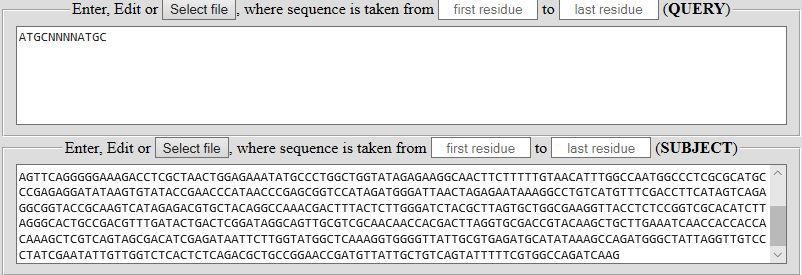
Find the occurrence or the closest analogue of the short biotechnological pattern ATGCNNNNATGC (QUERY) in the long nucleotide sequence (SUBJECT).
To solve the problem, it is necessary to take into account the following settings in the program:
| Sequences can be typed, copied or entered from a FASTA file. Random sequences can be generated and / or modified using individual services. | |
 |
|
| The program menu contains the switch of settings panel. By default, the program is consistent with the BLAST Global Alignment program. |  |
| Results include identity statistics, alignment score, performance measurement (times: total, of forward and reverse passes), alignment string with coordinates. When aligning two random sequences with default settings, close to 50% match is realized. | |
 |
|
| The choice of preset program settings is carried out using the drop-down menu. |  |
| In the scoring matrix positive values can be set for the mismatch of a nucleotide N with any other (the last row and the last column of the matrix) - that allows not to break the constriction NNNN. Another way to prohibit the break sequence is listed in the table below. |  |
| When aligning a short sequence to a long one, you need to set penalties for end inserts equal to 0 - which allows you to get rid of the influence of the ends on the result. | 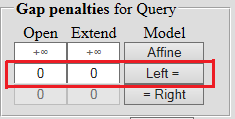 |
| To prohibit the break in the sequence or insert more than one character, you can set an infinite score, leaving the corresponding field empty, in which the infinity sign appears after that. | 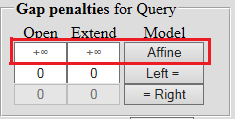 |
| The systems of priority in reverse pass can be changed or set random - that allows one to find all the options for the occurrence of the pattern. | 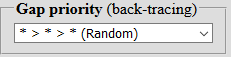 |
| If there are many cores on a computer, then you can set the desired number of processors (4 by default). The division of sequences into subsequences is carried out automatically, but can be set manually. | 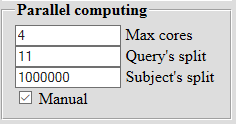 |